An overview of Cystic Fibrosis link
What is Cystic Fibrosis link
Cystic fibrosis (CF) is a genetic (inherited) disease that causes sticky, thick mucus to build up in organs, including your lungs and pancreas.
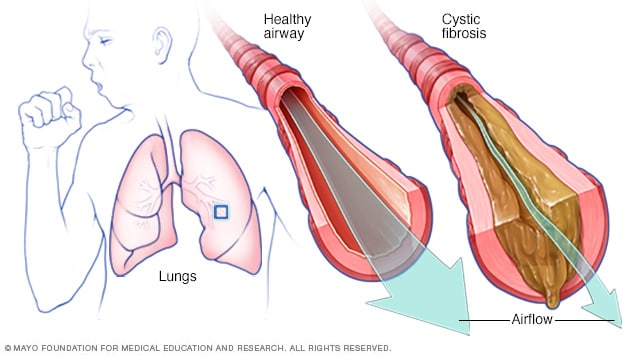
Over time, this thick mucus builds up inside your airways. This makes it hard to breathe. The mucus traps germs and leads to infections. It can also cause severe lung damage like cysts (fluid-filled sacs) and fibrosis (scar tissue). That’s how CF got its name.
Causes of Cystic Fibrosis link
Cystic fibrosis is caused by a change, or mutation, in a gene called CFTR (cystic fibrosis transmembrane conductance regulator). This gene controls the flow of salt and fluids in and out of your cells. If the CFTR gene doesn’t work the way it should, a sticky mucus builds up in your body.
Children need to inherit one copy of the gene from each parent in order to have the disease. If children inherit only one copy, they won't develop cystic fibrosis. However, they will be carriers and could pass the gene to their own children.
Signs and symptoms of Cystic Fibrosis link
Cystic fibrosis signs and symptoms vary, depending on the severity of the disease. Even in the same person, symptoms may worsen or improve as time passes. Some people may not experience symptoms until their teenage years or adulthood. People who are not diagnosed until adulthood usually have milder disease and are more likely to have atypical symptoms, such as recurring bouts of an inflamed pancreas (pancreatitis), infertility and recurring pneumonia.
People with cystic fibrosis have a higher than normal level of salt in their sweat. Parents often can taste the salt when they kiss their children. Most of the other signs and symptoms of CF affect the respiratory system and digestive system.
Respiratory signs and symptoms
The thick and sticky mucus associated with cystic fibrosis clogs the tubes that carry air in and out of your lungs. This can cause signs and symptoms such as:
~ A persistent cough that produces thick mucus (sputum)
~ Wheezing
~ Exercise intolerance
~ Repeated lung infections
~ Inflamed nasal passages or a stuffy nose
~ Recurrent sinusitis
Digestive signs and symptoms
The thick mucus can also block tubes that carry digestive enzymes from your pancreas to your small intestine. Without these digestive enzymes, your intestines aren't able to completely absorb the nutrients in the food you eat. The result is often:
~Foul-smelling, greasy stools
~Poor weight gain and growth
~ Intestinal blockage, particularly in newborns (meconium ileus)
~ Chronic or severe constipation, which may include frequent straining while trying to pass stool, eventually causing part of the rectum to protrude outside the anus (rectal prolapse)
Cystic Fibrosis Link Diagnosis
Early diagnosis means early treatment and better health later in life. Every state in the U.S. tests newborns for cystic fibrosis using one or more of these three tests:
Blood test. : This test checks the levels of immunoreactive trypsinogen (IRT). People with CF have higher levels of it in their blood.
DNA test : This looks for mutations to the CFTR gene.
Sweat test : It measures the salt in your sweat. Higher than normal results suggest CF.
Some people who weren’t tested at birth aren’t diagnosed with CF until they become adults. Your doctor might give you DNA or sweat tests if you have symptoms of the disease.
How is Cystic fibrosis Link treated?
You’ll probably have a healthcare team that includes a specialist in cystic fibrosis and many other types of caregivers. There is no cure for cystic fibrosis, but your team will help you manage the disease. The major focus of management is keeping your airways clear. Your provider will also prescribe medicine when needed.
Keeping airways clear
You can help to keep your airways clear if you have cystic fibrosis in a number of ways:
1. You can learn something called chest physical therapy, also known as postural drainage and percussion to loosen mucus. With this method, you move into certain positions so that your lungs can drain. Another person claps their hand on your chest and/or your back to help loosen the mucus. You might combine this with coughing.
2. You can learn special ways of coughing and breathing.
3. You can use devices that fit into your mouth or therapy vests that rely on vibrations to loosen mucus.
Medications for cystic fibrosis
Your provider may prescribe these medicines, which won’t cure CF, but which will help you in certain situations. They include:
1. Medications to treat the cause of cystic fibrosis in people with certain gene variants.
2. Anti-inflammatory drugs, including steroids and non-steroidal anti-inflammatories.
3. Antibiotics to treat lung infections or prevent them.
4. Inhaled bronchodilators to make breathing easier by opening and relaxing your airways.
5. Inhaled medicine to make mucus thinner and easier to get rid of.
6. Stool softeners to help with constipation.
7. Pancreatic enzymes to aid in digestion.
Surgeries for cystic fibrosis link
You may need surgery for cystic fibrosis or one of its complications. These might include:
1. Transplant surgery, including a double lung transplant or a liver transplant.
2. Surgery on your nose or sinuses.
3. Bowel surgery to remove blockages.
How can I prevent cystic fibrosis Link?
You can’t prevent cystic fibrosis because it’s an inherited condition. If you or your partner have any kind of family history, you may want to speak to a genetic counselor before you decide to have children.
What are the complications of cystic fibrosis Link?
The complications of CF include the following:
Adults who have CF can have problems with breathing, digestion and their reproductive organs.
The thick mucus present in people who have CF can hold bacteria, which can lead to more infections.
People who have CF have a higher risk of developing diabetes or the bone-thinning conditions like osteopenia and osteoporosis.
Men who have CF are not able to father children without the aid of alternative reproductive technology. Women who have CF can have a decrease in fertility (the ability to have children) and complications in pregnancy.
Frequently Asked Questions About cystic fibrosis Link
No. Cystic fibrosis isn’t contagious. It’s a genetic disorder, not an infection. You can’t catch it from anyone and you can’t give it to anyone. If you have it, though, you need to be careful if you’re exposed to infections.
Most cases of cystic fibrosis are found during the first few years of life. However, it’s possible to become an adult and then be diagnosed as having CF.
In the past, cystic fibrosis was considered to be a fatal illness. People who had it died in childhood. This is no longer true. Today, most children who have CF grow up to be adults with CF.
Among white children in the U.S., the rate of CF cases is 1 in 2,500 to 3,500 newborns. CF affects about 1 in 17,000 Black newborns and 1 in 31,000 newborns of Asian descent.